
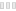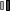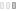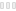
Случай из практики: болезнь Канавана
Невмержицкая К.С., Никитина Н.В., Сулимов А.В.
УДК 616.1: 616-053.2
ГОУ ВПО Уральская государственная медицинская академия Росздрава, г. Екатеринбург
Резюме.
Болезнь Канавана — это редкое аутосомно-рецессивное заболевание, связанное с мутацией гена аспартоацилазы. Результатом дефицита этого фермента является накопление в центральной нервной системе (ЦНС) N-ацетил-L-аспарагиновой кислоты (NAA) и дефицит ацетата, что приводит к формированию губчатой дегенерации белого и серого вещества головного и спинного мозга. Клинически заболевание проявляется макроцефалией, атрофией зрительных нервов, мышечной гипотонией, сменяющейся спастичностью, судорогами и грубой задержкой развития. Приводится клинический случай ранней диагностики болезни Канавана с типичным развитием заболевания.
Ключевые слова:
болезнь Канавана, N-ацетиласпартат
Развитие большинства лейкодистрофий связано с гипо— или дисмиелинопатиями, возникающими в результате первичного генетического дефекта синтеза миелина или вследствие вторичного влияния на изначально нормально протекающую миелинизацию нервных волокон [8]. В любом случае указанные нарушения влекут за собой тяжелую аксональную дисфункцию, клинические признаки которой проявляются уже в раннем детском возрасте. В этой статье мы представляем современные взгляды на этиологию, механизмы развития и коррекции одной из форм лейкодистрофий — болезни Канавана, так же известной как болезнь Канавана-Ван Богарта-Бертрана или спонгиозная дегенерация центральной нервной системы. Кроме того, мы приводим клиническое наблюдение подтвержденного случая болезни Канавана с типичным развитием заболевания.
Первое описание болезни было представлено в 1931 году Myrtelle Canavan, именем которого и стали называть заболевание [10]. В настоящее время известно, что болезнь Канавана — это редкое генетически детерминированное заболевание с аутосомно-рецессивным типом наследования. Оно встречается повсеместно с увеличением распространенности в популяции евреев-ашкенази, где частота его гетерозиготного носительства составляет 1:40-1:60 [12].
Развитие заболевания связано с дефектом структурного гена ASPA, ответственного за синтез фермента аспартоацилазы (аминоацилазы-2) и экспрессирующегося в олигодендроцитах, печени, скелетных мышцах, фибробластах кожи. В 1994 году в Майами было выяснено, что ген аспартоацилазы включает в себя 6 экзонов и локализован на 17 хромосоме. Позднее с помощью метода флюоресцентной гибридизации in situ было уточнено место расположения гена — 17pter-p13 [7]. К настоящему моменту известно не менее 12 мутаций в гене ASPA, встречающихся в популяции с различной частотой. Более всего распространены точковые мутации по типу нуклеотидных замен. При этом показано, что в еврейской популяции Ашкенази подавляющее большинство случаев (85-98%) болезни Канавана связано с мутациями p.Glu285Ala и p.Tyr231X. Среди европейцев повсеместно встречается мутация p.Ala305Glu в 6 экзоне гена ASPA (40-60% аллелей), которая, по мнению некоторых исследователей, является наиболее древней [12]. Значительно реже по сравнению с точковыми заменами в рассматриваемом гене описаны сплайсинговые мутации, делеции нуклеотидов и целых экзонов. В 2006 году было идентифицировано наличие компаунд-гетерозиготности при болезни Канавана, когда у пациентов на гомологичных хромосомах присутствовали 2 разных аллеля мутантного гена. Авторы описывали более «мягкую» или атипичную клиническую картину у носителей такой мутации: увеличение продолжительности жизни, отсутствие грубого двигательного дефекта и тяжелой задержки развития [5].
В 1988 году Matalon et al., обследуя семьи с болезнью Канавана, вывили значительное повышение концентрации N-ацетил-L-аспарагиновой кислоты в моче больных, чего не бывает при других лейкодистрофиях [10]. Таким образом, было сделано предположение о механизме развития заболевания — формирование метаболического блока на пути расщепления N-ацетиласпартата (NАА). Мутация гена ASPA приводит к снижению активности его продукта — фермента аспартоацилазы, в норме гидролизующей N-ацетил-L-аспарагиновую кислоту на L-аспартат и ацетат. В результате имеют место 2 патофизиологических эффекта — неконтролируемое накопление NАА в ЦНС с одной стороны и дефицит ацетата и аспарагиново й кислоты — с другой.
NАА и в норме выявляется в больших количествах в ЦНС (10 ммоль/г) и, по мнению ряда исследователей, является важным показателем нейрональной жизнеспособности [13]. Однако физиологическая роль NАА до конца не ясна. По одному из предположений, NАА активно вовлекается в нейрональную осморегулцию, и увеличение ее концентрации влечет за собой повреждение миелиновых оболочек и развитие хронического отека [11]. Предполагается так же токсическое действие N-ацетил-L-аспарагиновой кислоты на ткань мозга [1].
В развивающемся мозге NАА — источник ацетата для синтеза в олигодендроцитах ацетил-КоА, жирных кислот и в последующем — липидов мозга. В эксперименте показано, что снижение уровня ацетата в результате блокирования расщепления NАА приводит к снижению количества галактоцереброзидов в ЦНС [2]. Еще одна физиологическая роль ацетата состоит в ацетилировании гистоновых белков хроматина в ядрах олигодендроцитов [2]. Ацетилирование гистонов имеет ведущее значение в эпигенетическом контроле созревания олигодендроцитов, что в случае его нарушения из-за дефицита аспартоацилазы препятствует нормальной дифференцировке этих клеток, дисмиелинизации и сопровождается вакуолизацией серого и субкортикального белого вещества головного и спинного мозга.
Таким образом, вышеуказанные патофизиологические механизмы неуклонно приводят к отеку и губчатой дегенерации белого и серого вещества. При электронной микроскопии выявляют набухшие астроциты с вакуолизированной цитоплазмой и измененными митохондриями [1].
Для болезни Канавана характерно существование возрастного континуума, то есть дебют заболевания приходится на различные возрастные периоды, что позволяет выделить неонатальную, инфантильную и позднюю форму болезни. Наиболее распространена форма с дебютом в 3-5 месяцев после короткого промежутка относительно нормального развития. В первые месяцы дети могут демонстрировать элементы социального поведения — улыбаться, кратковременно гулить, реагировать на контакт. Однако в дальнейшем эти навыки постепенно утрачиваются. Довольно рано возникает атрофия зрительных нервов, что приводит к слабовидению. Типичным проявлением является макроцефалия. Важной особенностью в двигательной сфере следует считать диффузную мышечную гипотонию, которая постепенно трансформируется в спастичность. В первые несколько месяцев еще сохраняются произвольные движения, однако с течением заболевания снижается контроль положения головы, прогрессирует спастический тетрапарез, гиперрефлексия, патологические установки конечностей. С возрастом утяжеляются псевдобульбарные нарушении с тяжелой дисфагией, нередко требующей перехода на зондовое кормление. Нередко развитие заболевания сопровождается нарушением сна, возбудимостью, снижением порога действия раздражителей («стартл-рефлексы»); появляются судороги. Течение заболевания прогрессирующее, резкие ухудшения состояния провоцируются интрекуррентными инфекциями, переохлаждением. Продолжительность жизни от 3 до 10 лет [1, 8, 10].
Среди дополнительных методов диагностики ведущее значение приобретает магнитно-резонансная томография (МРТ), с помощью которой визуализируется довольно характерная картина диффузных симметричных очагов демиелинизации в Т2-режиме в подкорковых областях и в коре. Вовлечение мозжечка и ствола наблюдается реже [6]. На ранних этапах развития заболевания изменения могут не иметь столь выраженного характера, что требует очень тонкой дифференцировки с лейкоэнцефалопатией перинатального, инфекционного или иного генеза. Довольно информативным исследованием является протонная магнитно-резонансная спектроскопия головного мозга, выявляющая повышенную концентрацию NАА в центральной нервной системе [6, 13].
Уточняющими методами диагностики болезни Канавана являются биохимическая и генодиагностика. Наиболее распространенным подходом биохимической диагностики является выявление повышенного содержания N-ацетил-L-аспарагиновой кислоты в моче хроматографическими или масс-спектрометрическими методами. В норме ее содержание не превышает 700 мМ/М креатинина. При болезни Канавана концентрация NАА в моче увеличивается в 20 и более раз. В некоторых случаях прибегают к измерению активности фермента аспартоацилазы в фибробластах кожи [1].
К настоящему времени не накоплено достаточно данных, подтверждающих наличие эффективного метода лечения болезни Канавана. Однако это первое заболевание из всех лейкодистрофий, при котором была предпринята попытка генотерапии [8]. Двадцати одному пациенту с болезнью Канавана нейрохирургическим методом осуществили перенос нормального гена ASPA с помощью вирусного вектора в центральную нервную систему. Результаты проведенных исследований оказались противоречивыми. Наряду с выраженным увеличением активности аспартоацилазы и снижением уровня NАА двигательные улучшения не были столь значимыми, а при проведении нейровизуализации сохранялись явления губчатой дегенерации [4, 9]
Вполне перспективными считаются методы патогенетической терапии. В эксперименте показано, что препараты лития при болезни Канавана, снижают уровень NАА в ЦНС [3]. Шести пациентам в течение 6 месяцев вводили цитрат лития. По данным анкетирования родителей, больные стали более эмоциональными, а на МР-томограммах было отмечено появление миелинизации в белом веществе лобных долей. Двигательные нарушения оставались без изменений.
На основании утверждения, что явления дисмиелинопатии при болезни Канавана связаны с дефицитом ацетата, возникающим в результате блока гидролиза NАА, группой ученых был предложен метод заместительной терапии глицерилацетатом [2]. Показателями эффективности такой терапии стали расширение моторных навыков, увеличение содержания галактоцереброзидов и незначительное уменьшение спонгиформной вакуолизации белого вещества ЦНС. Исследования в области применении глицерилацетата продолжаются и направлены на поиск адекватной дозировки, способа и времени введения препарата, а так же на оценку его переносимости и безопасности.
Приводим собственное клиническое наблюдение случая болезни Канавана. Пациент М. мужского пола родился 5.07.2008 г. в семье русских родителей, не являющихся родственниками. Анте— и интранатальный периоды протекали без особенностей. С 1 месяца мать стала обращать внимание, что ребенок слабо фиксирует взор, не следит за предметами. На плановом осмотре офтальмологом глазного дна обнаружились признаки частичной атрофии зрительных нервов, что явилось поводом для дальнейшего обследования больного.
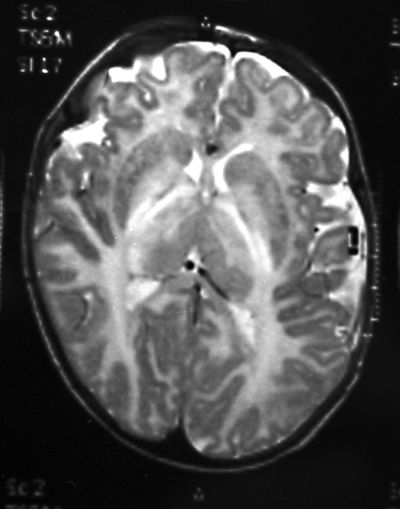
На осмотре обращали внимание беспокойство ребенка, оживление всех врожденных рефлексов. Определялась макроцефалия (динамика прироста окружности головы — 8 см за три месяца), отсутствие контроля положения головы и фиксации взора, мелкоразмашистый горизонтальный с ротаторным компонентом нистагм, псевдобульбарный синдром, пирамидный гипертонус в конечностях. Ребенок не улыбался, не гулил. При проведении магнитно-резонансной томографии головного мозга выявились зоны полного отсутствия миелинизации, в том числе в стволе и мозжечке, и признаки неполной миелинизации в зрительных нервах, перекресте, трактах и в наружном подкорковом белом веществе (рис.). Подобное распределение зон наличия и отсутствия миелинизации позволило предположить особый вид губчатой дегенерации белого вещества головного мозга — болезнь Канавана. Для подтверждения диагноза помимо клинических и нейровизуализационных признаков требовалось исследование биохимического фенотипа и проведение ДНК-диагностики. При исследовании концентрации N-ацетиласпарагиновой кислоты в моче методом тонкослойной хроматографии определялся ее повышенный уровень — 1463 мМ/М креатинина. Проведение ДНК-диагностики позволило выявить довольно частую мутацию в гене аспартоацилазы (ASPA) p.Ala305Glu в гомозиготном состоянии.
Дальнейшее течение заболевания представляло собой утяжеление нейродегенеративного регресса. Ребенок оставался малоэмоциональным и беспокойным. С 4-5 месяцев появился тремор и вздрагивания конечностей, провоцируемые минимальными раздражителями, как проявления стартл-реакций. В 6 месяцев дебютировали тонические короткие пароксизмы, сопровождающиеся эпилептиформной активностью на электроэнцефалограмме. Регулярное исследование неврологического статуса в динамике констатировало нарастание спастичности в конечностях со снижением объема активных движений, прогрессирующее нарушение глотания, опережение прироста окружности головы.
При осмотре в 1 год 3 месяца состояние тяжелое за счет неврологической симптоматики. Соматически компенсирован. На осмотр реагирует негативно в виде небольшого усиления двигательной активности. При тактильной стимуляции усиливается нистагм, рефлексы орального автоматизма, появляются множественные синкинезии. Окружность головы 55 см (более 97 центили). Со стороны черепной иннервации: взгляд не фиксирует, зрачки равны, фотореакции резко снижены, имеется преходящая установка на расходщееся косоглазие с двух сторон, грубый горизонтальный нистагм, асимметрия лица за счет сглаженности носогубной складки и опущения угла рта справа, псевдобульбарный синдром, девиация языка вправо. Спонтанная двигательная активность резко снижена, периодически отмечается дистоническая установка стоп. Мышечный тонус повышен по преимущественно пирамидному типу, существенно больше справа. Сухожильные рефлексы высокие, с расширением рефлексогенных зон; поверхностные кожные рефлексы снижены. Обращают внимание во время осмотра патологические синкинезии с рук и ног, обилие атипичных рефлексов. Ребенок самостоятельно не удерживает голову, не переворачивается, навыки вертикализации отсутствуют. Узнает голос матери, прислушивается, успокаивается на ее руках, не гулит.
Наследственные нейродегенеративные заболевания в практике невролога встречаются редко. Вот почему так важно определить диагностическую тактику в нужном направлении. Данный клинический случай демонстрирует довольно «простой» диагностический путь, когда однозначное толкование картины МРТ в совокупности с особенностями клинических проявлений являются решающими в назначении дальнейшей уточняющей диагностики. Это особенно важно для заболеваний с разработанными методами патогенетической терапии, когда во время поставленный диагноз определяет раннее начало эффективного лечения. В представленном же случае идентификация мутации в гене ASPA дает возможность проведения пренатальной диагностики последующих беременностей в этой семье.
Литература
1. Краснопольская К.Д. Наследственные болезни обмена веществ [Текст]. М.: Изд-во ГУ НИИ общей патологии и патофизиологии РАМН, 2005. — С.172-173.
2. Arun P., Madhavarao C.N., Moffett J.R. et al. Metabolic acetate therapy improves phenotype in the tremor rat model of Canavan disease. J Inherit Metab Dis. 2010 June; 33(3): 195—210.
3. Assadi M., Janson C., Wang D.J. et al. Lithium citrate reduces excessive intra-cerebral N-acetyl aspartate in Canavan disease. Eur J Paediatr Neurol. 2010 Jul;14(4):354-9.
4. Janson C, McPhee S, Bilaniuk L et al. Clinical protocol. Gene therapy of Canavan disease: AAV-2 vector for neurosurgical delivery of aspartoacylase gene (ASPA) to the human brain. Hum Gene Ther. 2002 Jul 20;13(11):1391-412.
5. Janson C.G., Kolodny E.H., Zeng B.-J. et al. Mild-onset presentation of Canavan‘s disease associated with novel G212A point mutation in aspartoacylase gene. Ann. Neurol. 2006; 59: 428-431.
6. Janson C.G., McPhee S.W., Francis J. et al. Natural history of Canavan disease revealed by proton magnetic resonance spectroscopy (1H-MRS) and diffusion-weighted MRI. Neuropediatrics. 2006 Aug;37(4):209-21.
7. Kaul R., Balamurugan K., Gao G. P., Matalon R. Canavan disease: genomic organization and localization of human ASPA to 17p13-ter and conservation of the ASPA gene during evolution. Genomics 1994; 21: 364-370.
8. Kumar S., Mattan N.S., de Vellis J. Canavan disease: a white matter disorder. Ment Retard Dev Disabil Res Rev. 2006;12(2):157-65.
9. Leone P., Janson C.G., Bilaniuk L. et al. Aspartoacylase gene transfer to the mammalian central nervous system with therapeutic implications for Canavan disease. Ann Neurol. 2000 Jul; 48(1): 27-38.
10. Matalon R., Michals K., Sebesta D. et al. Aspartoacylase deficiency and N-acetylaspartic aciduria in patients with Canavan disease. Am. J. Med. Genet. 1988; 29: 463-471.
11. Moffett J.R., Ross B., Arun P., Madhavarao C.N., Namboodiri A.M. N-Acetylaspartate in the CNS: from neurodiagnostics to neurobiology. Prog Neurobiol 2007; Feb; 81(2):89-131.
12. Sistermans E.A., de Coo R.F.M., van Beerendonk H.M., Poll-The B.T., Kleijer W.J., van Oost B.A. Mutation detection in the aspartoacylase gene in 17 patients with Canavan disease: four new mutations in the non-Jewish population. Europ. J. Hum. Genet. 2000; 8: 557-560.
13. Xu Su, Yang J., Shen J. Measuring N-acetyl aspartate synthesis in vivo using proton magnetic resonance spectroscopy. J Neurosci Methods. 2008 July 15; 172(1): 8—12.
Авторская справка
Невмержицкая Кристина Сергеевна
ГОУ ВПО Уралская государственная медицинская академия Росздрава
Россия, 620013, г. Екатеринбург, ул. Репина, д.3
e-mail: nks16@list.ru
Никитина Наталья Викторовна
СОГУЗ Центр планирования семьи и репродукции человека, г. Екатеринбург
Россия, 620000, г. Екатеринбург, ул. Флотская, д.52
Сулимов Алексей Валентинович
МУ Детская городская клиническая больница № 9
Россия, г. Екатеринбург, ул. Решетская, 51
e-mail: sulimov@e1.ru